Reaction Details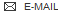 Report a problem with these data
Report a problem with these data
Report a problem with these dataTarget
Inhibitor of nuclear factor kappa-B kinase subunit beta
Ligand
BDBM50192071
Substrate
n/a
Meas. Tech.
ChEMBL_404533 (CHEMBL910916)
IC50
>1000±n/a nM
Citation
 Kawanishi, N; Sugimoto, T; Shibata, J; Nakamura, K; Masutani, K; Ikuta, M; Hirai, H Structure-based drug design of a highly potent CDK1,2,4,6 inhibitor with novel macrocyclic quinoxalin-2-one structure. Bioorg Med Chem Lett 16:5122-6 (2006) [PubMed] Article
Kawanishi, N; Sugimoto, T; Shibata, J; Nakamura, K; Masutani, K; Ikuta, M; Hirai, H Structure-based drug design of a highly potent CDK1,2,4,6 inhibitor with novel macrocyclic quinoxalin-2-one structure. Bioorg Med Chem Lett 16:5122-6 (2006) [PubMed] Article More Info.:
Target
Name:
Inhibitor of nuclear factor kappa-B kinase subunit beta
Synonyms:
I-kappa-B Kinase 2 (IKK-beta) | I-kappa-B kinase 2 | I-kappa-B-kinase beta | I-kappa-B-kinase beta (IKKB) | IKBKB | IKK-B | IKK-beta | IKK2 | IKK2/IKK1 | IKKB | IKKB_HUMAN | Inhibitor of NF-kappa-B kinase alpha/beta | Inhibitor of nuclear factor kappa B kinase beta subunit | NFKBIKB | Nuclear factor NF-kappa-B inhibitor kinase beta
Type:
Serine/threonine-protein kinase
Mol. Mass.:
86554.39
Organism:
Homo sapiens (Human)
Description:
GST-tagged IKK-2 was expressed in High Five cells and purified.
Residue:
756
Sequence:
MSWSPSLTTQTCGAWEMKERLGTGGFGNVIRWHNQETGEQIAIKQCRQELSPRNRERWCLEIQIMRRLTHPNVVAARDVPEGMQNLAPNDLPLLAMEYCQGGDLRKYLNQFENCCGLREGAILTLLSDIASALRYLHENRIIHRDLKPENIVLQQGEQRLIHKIIDLGYAKELDQGSLCTSFVGTLQYLAPELLEQQKYTVTVDYWSFGTLAFECITGFRPFLPNWQPVQWHSKVRQKSEVDIVVSEDLNGTVKFSSSLPYPNNLNSVLAERLEKWLQLMLMWHPRQRGTDPTYGPNGCFKALDDILNLKLVHILNMVTGTIHTYPVTEDESLQSLKARIQQDTGIPEEDQELLQEAGLALIPDKPATQCISDGKLNEGHTLDMDLVFLFDNSKITYETQISPRPQPESVSCILQEPKRNLAFFQLRKVWGQVWHSIQTLKEDCNRLQQGQRAAMMNLLRNNSCLSKMKNSMASMSQQLKAKLDFFKTSIQIDLEKYSEQTEFGITSDKLLLAWREMEQAVELCGRENEVKLLVERMMALQTDIVDLQRSPMGRKQGGTLDDLEEQARELYRRLREKPRDQRTEGDSQEMVRLLLQAIQSFEKKVRVIYTQLSKTVVCKQKALELLPKVEEVVSLMNEDEKTVVRLQEKRQKELWNLLKIACSKVRGPVSGSPDSMNASRLSQPGQLMSQPSTASNSLPEPAKKSEELVAEAHNLCTLLENAIQDTVREQDQSFTALDWSWLQTEEEEHSCLEQAS
Inhibitor
Name:
BDBM50192071
Synonyms:
(13R,15S)-13-methyl-16-oxa-8,9,12,22,24-pentaazahexacyclo[15.6.2.1^{6,9}.1^{12,15}.0^{2,7}.0^{21,25}]heptacosa-1(24),2,4,6(27),7,17(25),18,20,22-nonaene-23,27-diol | CHEMBL215803
Type:
Small organic molecule
Emp. Form.:
C22H21N5O3
Mol. Mass.:
403.4338
SMILES:
C[C@@H]1C[C@H]2CN1CCn1[nH]c3c(cccc3c1=O)-c1nc3c(O2)cccc3[nH]c1=O
Structure: